
Einblick in die dunkle Materie des Genoms
Archivmeldung vom 22.11.2019
Bitte beachten Sie, dass die Meldung den Stand der Dinge zum Zeitpunkt ihrer Veröffentlichung am 22.11.2019 wiedergibt. Eventuelle in der Zwischenzeit veränderte Sachverhalte bleiben daher unberücksichtigt.
Freigeschaltet durch Manuel Schmidt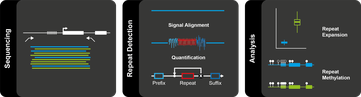
DNA-Sequenzwiederholungen können zu Krankheiten führen, lassen sich aber kaum untersuchen. Ein Verfahren von Forschenden des Max-Planck-Instituts für molekulare Genetik ermöglicht erstmals einen detaillierten Blick auf diesen zuvor unzugänglichen Bereich des Genoms. Dazu kombiniert es Nanopore-Sequenzierung, Stammzelltechnologie und CRISPR-Cas. Das Verfahren könnte die Diagnostik von verschiedenen angeborenen Erkrankungen und Krebserkrankungen verbessern. Die Ergebnisse erschienen in Fachblatt Nature Biotechnology.

Weite Teile des Genoms bestehen aus monotonen Bereichen, in denen sich kurze Abschnitte des Genoms hunderte oder tausende Male wiederholen. Solche „DNA-Repeats“ können manchmal dramatische Auswirkungen für die Betroffenen haben, zum Beispiel beim Fragilen-X-Syndrom, einer der häufigsten identifizierbaren erblichen Ursachen für kognitive Behinderungen des Menschen. Doch die repetitiven Regionen gelten noch als unbekanntes Terrain im Genom. Sie lassen sich auch mit modernen Verfahren nicht exakt untersuchen.
Wenn es nach dem Forschungsteam um PD Dr. Franz-Josef Müller am Max-Planck-Institut für molekulare Genetik in Berlin und am Universitätsklinikum Schleswig-Holstein in Kiel geht, soll sich das nun ändern. Das Team charakterisierte erstmals detailliert die Länge der Repeat-Sequenzen in Stammzellkulturen, die aus dem Gewebe von Erkrankten gewonnen wurden. Neben der Sequenzinformation gewannen die Wissenschaftlerinnen und Wissenschaftler auch Daten über den epigenetischen Zustand der Repeats anhand einzelner DNA-Moleküle. Damit stößt das Verfahren, das auf Nanopore-Sequenzierung und CRISPR-Cas-Technologien basiert, die Tür auf für die Erforschung von DNA-Repeats, sowie für die schnelle und genaue Diagnostik einer Reihe von Erkrankungen.
Gendefekt auf dem X-Chromosom
Beim Fragilen-X-Syndrom befindet sich der DNA-Repeat in einem Gen namens FMR1, das sich auf dem X-Chromosom befindet und für die Gehirnentwicklung von Bedeutung ist. „Die Zelle erkennt die wiederholten Bereiche und markiert sie durch das Anhängen von Methylgruppen als inaktiv“, sagt Müller. Trotz dieser kleinen chemischen Veränderungen bleibt die genetische Information erhalten, sie wirken also epigenetisch.
„Leider greift die epigenetische Markierung auf das ganze Gen über, das dadurch komplett abgeschaltet wird“, erklärt der Mediziner. „Das Deaktivieren des FMR1-Gens führt zu schweren Verzögerungen in der Entwicklung und zu unterschiedlich ausgeprägter Verminderung der Intelligenz oder zu Autismus.“
Das Syndrom gehört zu den etwa 30 Repeat-Erkrankungen, die beim Menschen bekannt sind. Frauen zeigen – wenn überhaupt – einen anderen Krankheitsverlauf, da sich die verlängerte Repeat-Region des FMR1-Gens meist nur auf einem der beiden X-Chromosom befindet. Die unveränderte, zweite Kopie des Gens bei Frauen funktioniert hingegen, trägt keine epigenetisch veränderte Signatur und kann somit den Gendefekt ausgleichen. Männer haben lediglich ein X-Chromosom und zeigen das volle klinische Bild des Syndroms.
Sequenz-Wiederholungen erstmals genau kartiert
Müller und sein Team untersuchten Stammzellen, die sie aus dem Gewebe von Erkrankten erzeugten. In den untersuchten Zellen konnten sie erstmals die Länge der Repeat-Regionen und deren epigenetische Signatur bestimmen. Dies ist mit herkömmlichen Sequenzierungsmethoden bislang nicht möglich gewesen. Sie entdeckten außerdem, dass die Region in den Zellen eines einzelnen Patienten sehr unterschiedlich lang sein kann.
Ihr Experiment wiederholten die Forscherinnen und Forscher an Zellen von Betroffenen mit einer völlig anderen Repeat-Erkrankung, die eine der häufigsten monogenen Ursachen für Frontotemporale Demenz und Amyotrophe Lateralsklerose darstellt. Hier liegt eine Sequenzwiederholung in einer Kopie des Gens C9orf72 vor. Die andere Genkopie ist dagegen intakt.
„Wir konnten erstmals die gesamte Epigenetik der verlängerten Repeat-Region im Vergleich zur unveränderten Repeat-Region in ein und demselben experimentellen Ansatz abbilden“, sagt Müller. Durch die Versuche sei der untersuchte DNA-Bereich völlig unberührt geblieben. „Wir haben eine bisher einzigartige Analysemethode für Einzelmoleküle und für die dunkelsten Regionen im Genom – das macht die Sache so spannend.“
Winzige Poren messen einzelne Moleküle
„Bei der Untersuchung von Repeat-Sequenzen stoßen konventionelle Sequenzierungs-Methoden an ihre Grenzen“, sagt Björn Brändl, einer der Erstautoren der Publikation. „Von der gleichzeitigen Detektion epigenetischer Eigenschaften von Repeats ganz zu schweigen.“ Das Forschungsteam verwendet daher die Nanopore-Technologie. Dabei wird die DNA zunächst in große Abschnitte zerlegt und jeder DNA-Einzelstrang durch eines von hunderten winzigen Löchlein („Nanopores“) auf einem Chip gefädelt.
Durch die Nanoporen strömen jederzeit elektrisch geladene Teilchen – es fließt also ein Strom. Bewegen sich nun DNA-Moleküle durch diese Poren, wird der Stromfluss je nach chemischer Zusammensetzung der vier unterschiedlichen DNA-Bausteine charakteristisch verändert. Aus Schwankungen in dem elektrischen Signal rekonstruiert der Computer anschließend die Abfolge der genetischen Buchstaben sowie ihre epigenetischen chemischen Markierungen – für jede einzelne Pore und damit jeden einzelnen DNA-Strang.
Genomeditierung und Bioinformatik erschließen „dunkle Bereiche“
Anders als bei konventionellen Sequenziermethoden, die das gesamte Erbgut einer Patientin bzw. eines Patienten analysieren, nutzte Brändl das CRISPR-Cas-System, das als hochpräzise „Gen-Schere“ bekannt ist. Der Wissenschaftler schnitt mit dem Werkzeug gezielt jene DNA-Stücke aus dem Erbgut heraus, für die sich das Team interessierte und die auch die Repeat-Region enthielten. Nur diese Segmente konnten dann nach weiteren Zwischenschritten anschließend von den Poren auf dem Sequenzier-Chip gelesen werden.
„Wenn wir die Moleküle nicht vorsortieren, geht ihr Signal im Rauschen des restlichen Genoms unter“, sagt der Bioinformatiker Pay Giesselmann. Er hat eigens für die Interpretation der elektrischen Signale einen Algorithmus entwickelt. „An den repetitiven Sequenzen scheitern herkömmliche Analyseprogramme, weil sie solche regelmäßigen Muster nicht erwarten.“ Sein Programm „STRique“ erstellt nicht die Abfolge der genetischen Buchstaben, sondern zählt die Anzahl der Sequenzwiederholungen. Es ist frei im Internet verfügbar.
Die Tür aufgestoßen für Forschung und Praxis
„Das CRISPR-Cas-System zusammen mit neuartigen Algorithmen ermöglicht es uns, jeden gewünschten Abschnitt des Erbguts gezielt unter die Lupe zu nehmen – insbesondere jene, die mit herkömmlichen Methoden nur schwer zu untersuchen sind,“ sagt der Studienleiter Müller. „Damit haben wir die Werkzeuge geschaffen, mit denen jeder die dunkle Materie des Genoms erschließen kann.“ Müller sieht in der neuen Methode großes Potential für die Grundlagenforschung. „Es wird vermutet, dass die Repeats während der Entwicklung des Nervensystems wachsen – das würden wir uns gern genauer ansehen.“
Auch für die Diagnostik ergeben sich kaum überschaubare Möglichkeiten, sagt der Mediziner. Schließlich seien Repeat-Regionen auch an der Entstehung von Krebs beteiligt und das Verfahren sei kostengünstig und schnell. Entsprechende Projekte seien bereits auf dem Weg: „Wir sind hier ganz nah an der klinischen Anwendung.“
Quelle: Max-Planck-Institut für molekulare Genetik (idw)




